
فهرست مطالب
محققان بهتازگی پلتفرمی ایجاد کردهاند که آزمایش های خودکار را با هوش مصنوعی ترکیب میکند و می تواند بینشی در مورد نحوه تعامل مواد شیمیایی برای ساخت دارو های مختلف ارائه دهد.
به گزارش تکناک، کشف و تولید داروهای جدید اغلب نیازمند پیشبینی دقیق واکنشهای مولکولی است. دانشمندان همیشه خاطرنشان میکنند که این فرآیند، یک فرآیند آزمون و خطای پر مشکل بوده است که اغلب منجر به واکنشهای ناموفق می شود. شیمیدانان به طور سنتی از شبیه سازی الکترونها و اتمها در مدل های ساده شده برای پیشبینی این واکنش ها استفاده میکنند، اما این روش از نظر محاسباتی گران است و مستعد عدم دقت است.
مدل جدید که توسط تیمی از محققان کالج دانشگاه کمبریج توسعه داده شده است، بر یک رویکرد داده محور و الهام گرفته از ژنومیک که در آن آزمایش های خودکار با یادگیری ماشینی برای درک واکنش شیمیایی ترکیب می شوند تا سرعت فرآیند ساخت دارو را تا حد زیادی بهبود بخشد، متکی است. طبق بیانیه محققان ادعا میشود که این سیستم که reactome شیمیایی نامیده می شود، با استفاده از مجموعه داده ای که شامل 39000 واکنش دارویی مرتبط است، آموزش داده می شود.
جزئیات تحقیقات با همکاری کمبریج و فایزر انجام شده است، در مجله Nature Chemistry منتشر شده است.
01
از 02سریع و قابل اعتماد
روش reactome همبستگیهای قابلتوجهی را بین واکنشدهندهها، معرفها و عملکرد کلی واکنشها در مجموعه داده انتخاب میکند و همچنین به کاستیهای موجود در خود داده اشاره میکند. دانشمندان خاطرنشان می کنند که اطلاعات تولید شده از آزمایشهای خودکار، سریع و قابل اعتماد است.
دکتر آلفا لی که این تحقیق را رهبری میکرد، گفت: رویکرد ما روابط پنهان بین اجزای واکنش و پیامدها را آشکار می کند. مجموعه داده ای که ما مدل را بر روی آن آموزش دادیم بسیار بزرگ است و به رساندن روند اکتشاف شیمیایی از آزمون و خطا به عصر دادههای بزرگ کمک می کند.
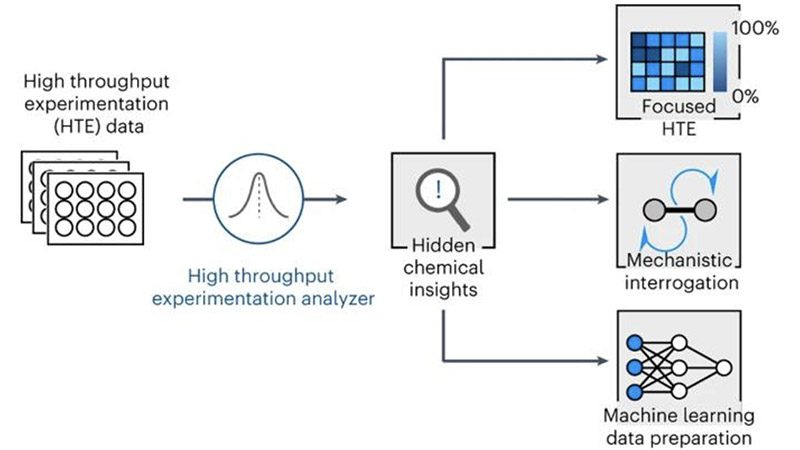
علاوه بر آزمایشهای خودکار، محققان یک روش یادگیری ماشینی ابداع کردهاند که شیمیدانان را قادر میسازد تا تغییرات دقیقی را در بخشهای مولکولی از پیش تعیینشده اعمال کنند و فرآیند طراحی دارو سریعتر را ارائه دهند. این رویکرد نوآورانه به شیمیدانان اجازه میدهد تا مولکولهای پیچیدهای را که شبیه اصلاحات لحظه آخری هستند، تنظیم کنند، بدون اینکه نیاز باشد از ابتدا شروع کنند. در محیط آزمایشگاهی سنتی، ساخت یک مولکول معمولاً شامل یک فرآیند چند مرحله ای شبیه به ساختن یک خانه است.
محققان خاطرنشان می کنند که وقتی شیمیدانها به دنبال اصلاح ساختار بنیادی یک مولکول هستند، روش مرسوم اساساً شامل از بین بردن کل مولکول و بازسازی مجدد آن است؛ چیزی شبیه به تخریب یک خانه و ساخت دوباره آن. با این وجود، ایجاد تغییرات هسته بدون بازسازی کامل در طراحی دارویی بسیار مهم است.
02
از 02برطرف کردن موانع
واکنشهای عاملسازی در مرحله آخر که به دسته خاصی از تبدیلهای شیمیایی تعلق دارند، با هدف اعمال تغییرات مستقیم در هسته یک مولکول، نیاز به شروع فرآیند را از ابتدا حذف میکنند. علیرغم مزایا، دستیابی به گزینش پذیری و کنترل در عملکردی سازی در مراحل آخر مشکلاتی را ایجاد می کند. محققان میگویند که مشکل اصلی از حساس بودن مناطق درون مولکولها به واکنش ناشی میشود که پیشبینی و کنترل نتیجه نهایی را دشوار میکند.
نویسنده اول این مطالعه، دکتر اما کینگ اسمیت گفت: عملکردی سازی در مراحل پایانی می تواند نتایج غیرقابل پیش بینی به همراه داشته باشد و روش های فعلی مدل سازی از جمله شهود تخصصی خودمان، بی نقص نیستند. استفاده از یک مدل پیش بینی کنندهتر فرصت غربالگری بهتر را به ما می دهد.
به همین منظور دانشمندان یک مدل یادگیری ماشینی ایجاد کردند که قادر به پیش بینی مکان خاص در یک مولکول است که در آن واکنش رخ می دهد. علاوه بر این، این مدل نحوه تغییر مکان واکنش تحت شرایط مختلف واکنش را پیشبینی میکند. این قابلیت به شیمیدانان کمک میکند تا روشهایی را برای تنظیم دقیق ساختار مرکزی یک مولکول با دقت بررسی کنند. کینگ اسمیت گفت: ما این مدل را بر روی حجم وسیعی از دادههای طیفسنجی از قبل آموزش دادیم.
با توجه به کمبود واکنشهای عاملسازی در مراحل آخر، این روش محققان را قادر میسازد تا به مشکلات مربوط به دادههای محدود رسیدگی کنند. محققان این مدل را به صورت تجربی با استفاده از مجموعهای متنوع از مولکولهای دارومانند تأیید کردند و با موفقیت مکانهای واکنشپذیری را در شرایط مختلف پیشبینی کردند.



















